

斑马鱼模型揭示遗传性线粒体疾病新致病机制
2020年11月23日,加州大学旧金山分校Gladstone 研究所Philipp Gut和Eric Verdin课题组、Buck老龄化研究所Birgit Schilling课题组、芬兰赫尔辛基大学Anu Suomalainen课题组合作在Nature Communications上在线发表了题为《SUCLA2 mutations cause global protein succinylation contributing to the pathomechanism of a hereditary mitochondrial disease》的研究论文,提出SUCLA2基因突变导致SCL缺乏,引起细胞内琥珀酰辅酶A积聚,进而引起全面的蛋白质过度酰基化,影响蛋白质的翻译后修饰过程,从而导致线粒体功能损伤的病理机制。此项工作中,研究人员采用斑马鱼模型揭示了SIRT5基因对蛋白琥珀酰化过程的调节作用,这一机制的发现将有助于该疾病治疗方法的开发。
线粒体内多种酰基辅酶A是蛋白质修饰和损伤的重要参与者。琥珀酰辅酶A连接酶(Succinyl-CoA ligase,SCL)缺乏是一种线粒体DNA耗竭综合症(OMIM#612073,#245400),患者表现出脑肌病特征。目前已报导超过20名患者携带SUCLG1(编码SCL α-亚基)突变,50名患者携带SUCLA2(编码ATP-特异性β-亚基)突变。携带SUCLA2突变的患者通常表现为儿童早期进行性脑肌病,包括张力减退、肌张力障碍和感音神经性耳聋。目前,SCL缺乏导致线粒体脑肌病的病理机制仍然未知。

研究者首先分析了携带SUCLA2突变的SCL缺乏患者体内的整体蛋白质琥珀酰化水平(图1)。利用非靶向代谢组学分析患者和对照组的成纤维细胞样本后,发现患者的琥珀酰辅酶 A水平显著上升。同时,SDS-PAGE和western blot结果显示患者的整体蛋白质琥珀酰化程度更高。这一结果表明SCL缺乏导致整体蛋白质过度琥珀酰化。
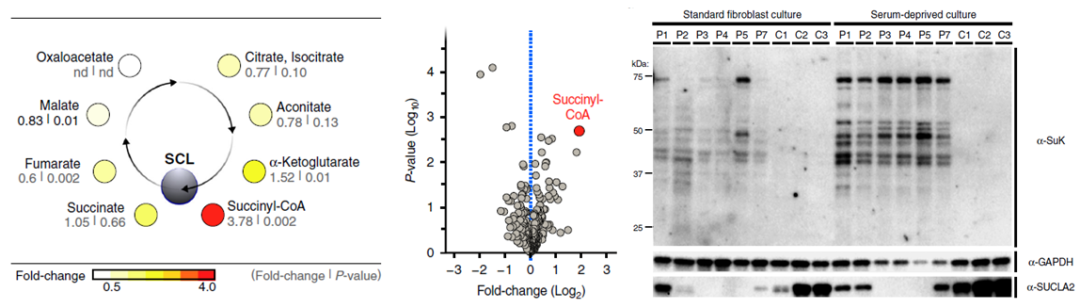
图1
SCL缺乏导致整体蛋白质过度琥珀酰化
接下来,通过对赖氨酸琥珀酰化靶点的蛋白组学分析,研究者在SUCLA2突变患者样本中检测到了更多的赖氨酸琥珀酰化位点(图2)。GO(Gene Ontology)富集分析显示参与三羧酸循环(Tricarboxylic acid cycle, TCA cycle)的蛋白质赖氨酸琥珀酰化位点富集程度最高,除此之外还包括ATP生物合成、丙酮酸代谢和β-氧化等代谢途径中参与线粒体能量生产的蛋白质。这表明SCL缺乏导致高水平的琥珀酰辅酶A产生,琥珀酰辅酶A又通过赖氨酸琥珀酰化影响不同细胞途径中的蛋白质。
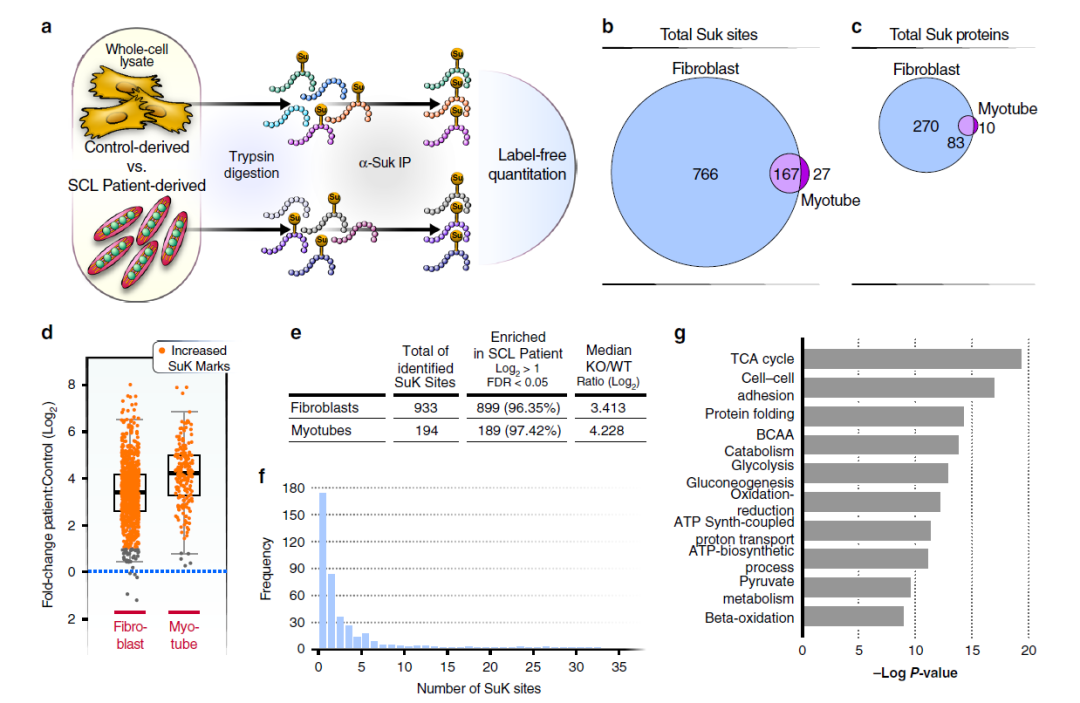
图2
赖氨酸琥珀酰化的蛋白组学分析
研究者还发现SUCLA2突变患者成纤维细胞模型中病理性琥珀酰化的蛋白质与Sirt5缺失小鼠模型中被显著调控的蛋白质大量重叠。为了探究SIRT5是否具有调节蛋白琥珀酰化的功能,研究者利用CRISPR/Cas9基因编辑技术在斑马鱼上进行了sirt5基因的功能研究。研究者首先建立了SCL缺乏斑马鱼模型(sucla2-/-),在此基础上分别建立sirt5功能缺失模型(sirt5-/-)和功能获得模型Tg(ubi:sirt5)。实验结果显示,仅sirt5功能缺失在幼鱼期不会导致蛋白的过度琥珀酰化,而sirt5和sucla2的共同缺失则会显著增加蛋白琥珀酰化水平,sirt5的功能获得模型可以部分减弱蛋白的过度琥珀酰化程度(图3)。这表明sirt5是病理性蛋白琥珀酰化过程的调节基因。
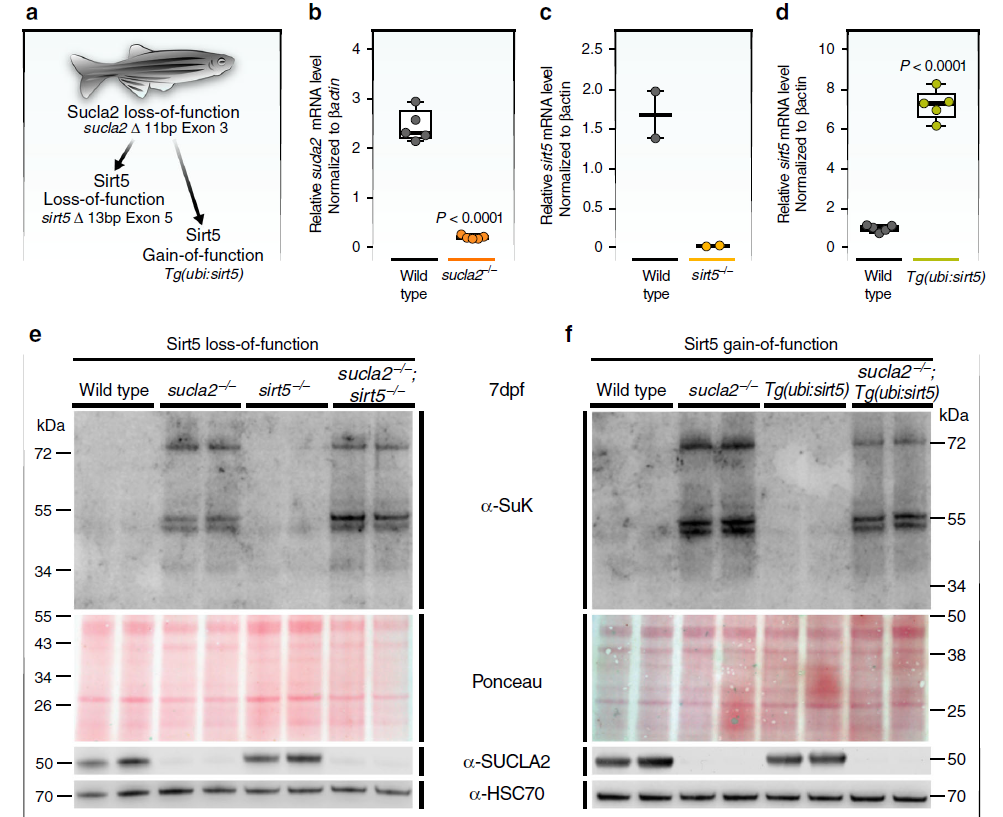
图3
SCL缺乏斑马鱼模型中,sirt5基因调控整体蛋白质琥珀酰化水平
为了解sirt5基因功能对SCL缺乏斑马鱼模型个体生存的影响,研究者测定了幼鱼的耗氧率(Oxygen consumption rate, OCR),发现sirt5基因过表达不能恢复线粒体DNA含量,但可显著提高sucla2缺失斑马鱼的OCR,并提高存活率(图4)。
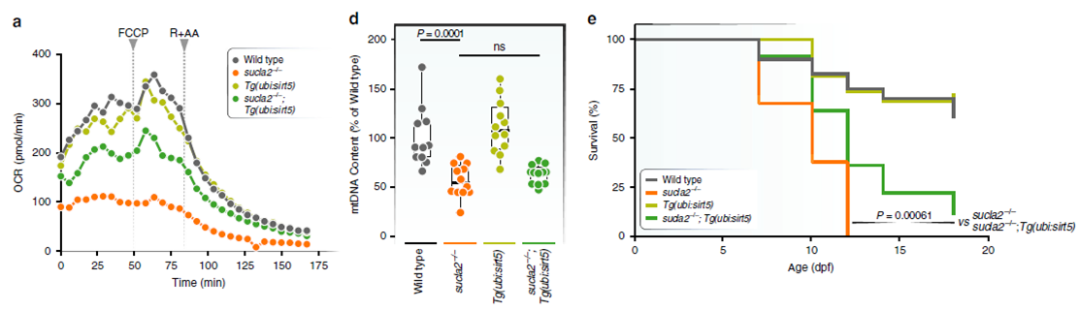
图4
SCL缺乏斑马鱼模型中,sirt5基因过表达可恢复幼鱼氧化代谢水平并提高存活率
综上所述,琥珀酰辅酶A在携带SUCLA2突变的SCL缺乏患者的细胞中积累,导致整体蛋白质的过度琥珀酰化,而sirt5功能增益可明显缓解SCL缺乏斑马鱼的蛋白过度琥珀酰化,恢复氧化代谢水平,提高整体生存率。
原文链接:https://doi.org/10.1038/s41467-020-19743-4