

转录因子PAX7基因的双等位变异是引起肌病的新的遗传原因
2019年5月16日的GENETICS in MEDICINE 杂志上发表了一篇名为《Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy》的文章。该文章通过对5个肌病家系的基因检测及相关功能验证,确认PAX7 基因的双等位变异是导致这一类肌病的新的遗传原因。

入组患者
五个肌病患者,分别来自四个血缘关系不相关的家庭。1号——14岁男孩,来自加拿大;2号——5岁女孩,来自德国;3号——男孩(7岁时死亡),来自巴勒斯坦;4号——4岁男孩,来自沙特阿拉伯;5号——6岁女孩,来自沙特阿拉伯。
入组患者临床表型:
五个患者除了2号患者在胎儿期表现出胎动减少异常外,其他患者均表现正常的胎儿期发育。
1号和2号患者出生即表现肌病表型,3、4和5号在围产期均表型正常。患者1在妊娠41周时出生,出生即发现吸吮较差,需要从出生时就用试管喂养。患者2在孕39周出生,出生即表现脓毒症、发绀,随后出现肌无力、血浆乳酸增加等线粒体能量代谢异常的常见疾病特征,但未检出乙酰胆碱抗体,代谢筛查呈阴性,也未见呼吸链酶活性受损迹象。
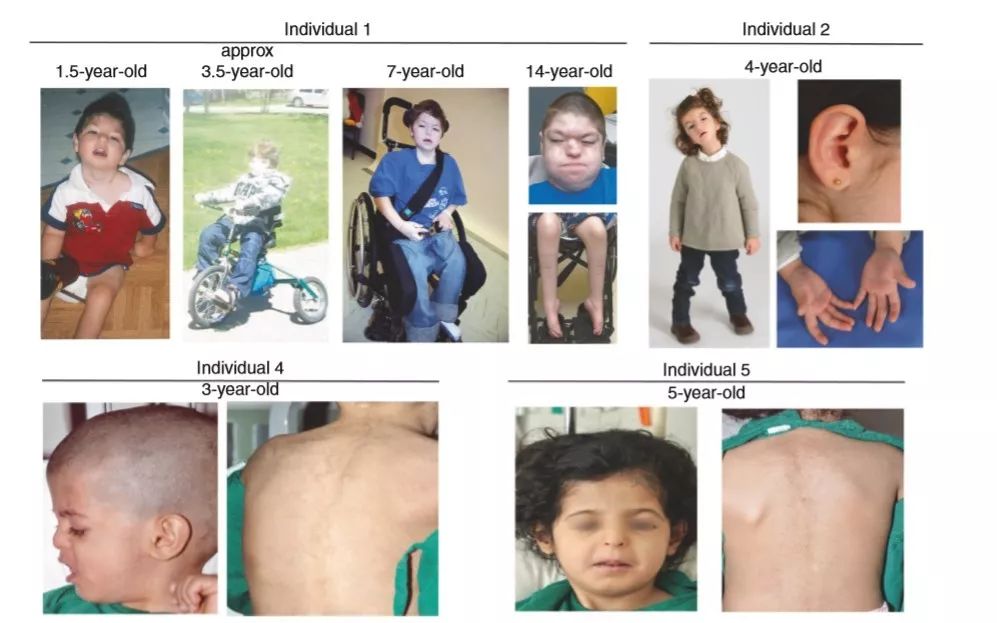
最初的临床症状在5个患者中均出现在出生后几个月,包括轴向或全身张力减退。每个人都观察到骨骼肌萎缩,主要影响躯干/颈部肌肉和/或下肢和远端肢体。所有的患者都表现生长对称,但是绝大多数患者都表现身材矮小或生长不足,且均表现严重不一的脊柱侧弯。绝大多数患者的步态受到影响:患者1——轮椅行走、患者3——行走困难、患者4——蹒跚行走、患者5——足尖和脚跟行走困难,但患者2被认为是正常的。
该肌病呈现随年龄进展的特点:年龄最大的1号患者在6岁前能够借助助行器行走,之后只能坐轮椅,现在14岁已经卧床不起。此外,由于肌肉无力引起的进行性呼吸功能不全症状也出现了,因此患者1开始采用双相气道正压通气(BiPAP)治疗,最初仅在夜间进行,后来需要持续进行。另外,在患者3中也观察到疾病严重程度的进展(他已经在7岁时死亡)。此外,脊柱侧弯的进展也在4号和5号患者中观察到,从2.7岁时的40°Cobb角到4岁时的60°Cobb角。
上睑下垂也是该类肌病常见的一个特征,除3号患者外,其他患者均表现此特征。
5个患者呈现可变的畸形面部特征,患者4和5表现孤立的三角形脸;患者1表现为长头、斜头畸形、小下颌,高弓腭,后旋耳,鼻根突出;患者2表现低耳位,低张相,高腭,扁平鼻,面部多毛。
5个患者均表现正常的认知和社交能力。
实验室检查
患者1的肌酸激酶(CK)、氨、血浆氨基酸、尿有机酸、转铁蛋白等电聚焦、脆性X染色体、核型、比较基因组杂交(CGH)芯片、强直性营养不良1型(MIM 160900)关联基因DMPK 基因分析等结果均表现阴性。其他患者的CK水平也均正常,表明没有肌肉退化/损伤。
患者1在3个月和2岁时肌电图(EMG)检查正常,但在13.5岁时检查出异常,提示有肌病。而其他患者肌电图检查都是正常的,表明这类疾病不是神经肌肉或神经病的起源。
5个患者的超声心动图和脑磁共振成像(MRI)结果均阴性。
肌肉活检分析
对患者1、2和4进行了肌肉活检检查。肌肉活检分析显示没有肌纤维变性的迹象;但卫星细胞的缺乏导致肌肉生长受损和纤维脂肪组织的替代,从而导致进行性肌无力。
基因检测
对5个患者进行全外显子组测序,结果显示5个患者在目前与肌病相关的已知基因中没有任何可疑罕见变异的发现。
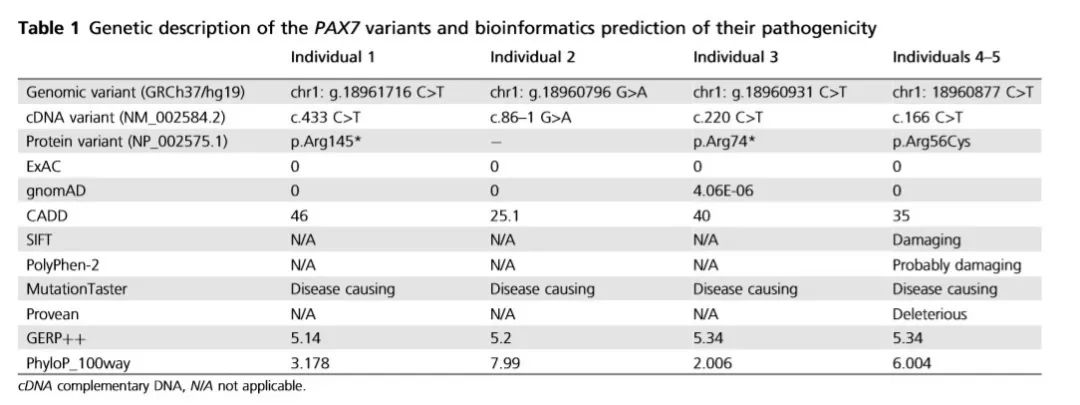
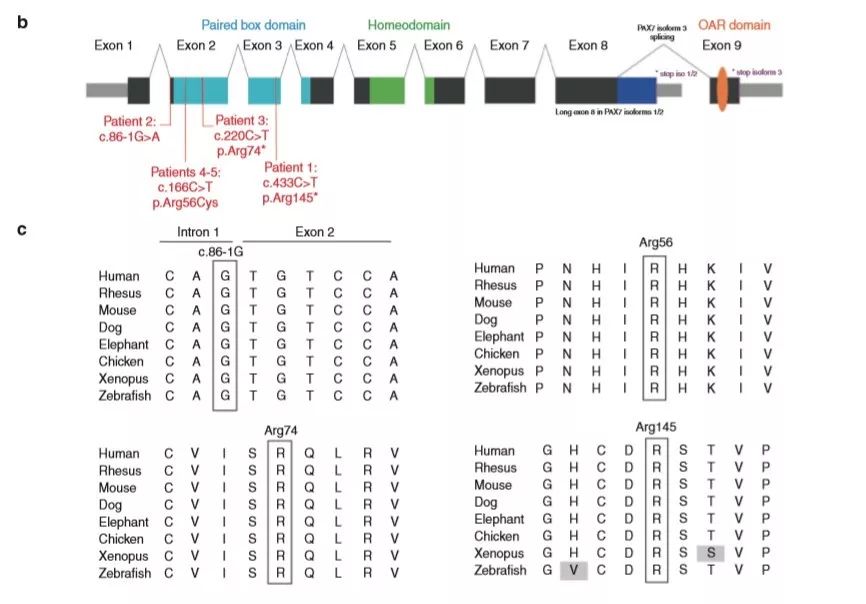
研究人员在常染色体隐性遗传的假设下,锁定了PAX7 基因——发现5个患者在PAX7 基因上分别存在4种不同类型的的罕见纯合变异,详见下表。这些变异在ExAC和gnomAD数据库中没有被收录或频率极低;且影响非常保守的氨基酸残基;此外,这些变异被多个软件预测有害。
PAX7 在普通人群中非常保守,且对功能缺失的变异非常不耐受。
肌源性调节因子的表达分析
取1号和2号患者的肌肉活检组织进行定量PCR检测。分析结果显示,与同龄对照组相比,患者1和患者2的PAX7 转录量显著下降,PAX7 基因的下游靶点MYF5 基因的表达水平也有类似的下降;同时免疫染色证实在患者1和患者2中完全缺失PAX7 表达和MYF5 表达的细胞。与PAX7 和MYF5 基因的表达结果相反,这两个患者的胚胎肌球蛋白重链3(MYH3 )基因表达均升高,提示肌纤维再生活跃,同时免疫染色实验表明表达MYH3 的肌纤维只在患者的肌肉中被观察到(在健康对照组中没有),表明存在正在进行的肌肉再生。
总结
PAX7 是Paired box 基因家族中的一员,这个家族的所有成员都共享一个高度保守的DNA结合域,即128个氨基酸的保守成对结构域。PAX 基因家族共有9个不同的基因,它们通过调节细胞分化和祖细胞的维持,在器官发生和组织发育中发挥重要作用。目前已有的研究发现除了基因PAX7,其他所有的PAX 家族基因的功能丧失变异都与遗传疾病有关。而PAX7 目前发现与疾病的关系是,在体细胞中PAX7与FOXO1 的基因融合会导致肺泡横纹肌肉瘤(IMI 268220)。PAX7 功能丧失对人体组织发育和病理的影响尚不清楚。在对PAX7 基因功能的研究中,曾有人构建PAX7 -/-小鼠模型,发现此模型小鼠在胚胎发育期间表现正常,但在断奶后不久即死亡。这些小鼠在出生时表现出上颌骨和鼻子等面部结构的畸形,但没有明显的中枢神经系统或骨骼肌的异常。研究发现,胚胎期神经系统和骨骼肌的发育主要依赖于PAX3 基因,而PAX7 是影响产后肌肉生长和再生的主要因素。PAX7 功能缺失的变异并不直接影响肌纤维结构和/或功能,而是导致卫星细胞池的衰竭。这项研究首次确定了影响肌肉干细胞的肌肉病变的遗传原因。
参考文献:
Rene G. Feichtinger, Betting E. Mucha, Holger Hengel, et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy[J]. Genet Med. 2019 May 16. doi: 10.1038/s41436-019-0532-z.