

赛福基因公开课《几种常见神经皮肤综合征相关疾病基因突变研究简介》

各位老师好,我是范珊珊。今天很荣幸和各位老师一起探讨一下与几种常见神经皮肤综合征相关的一些遗传学知识。
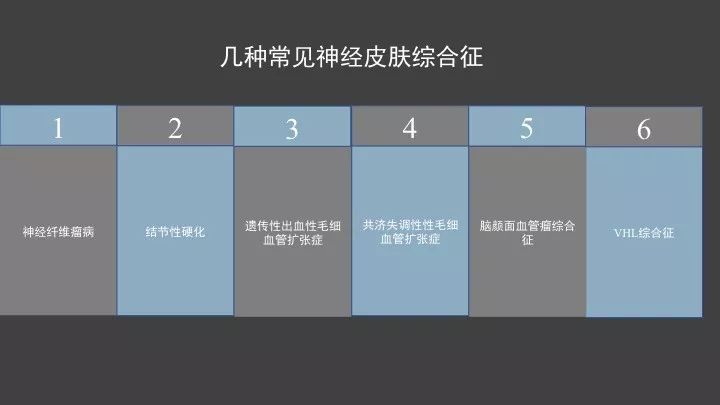
今天我们主要介绍6种临床中常见的神经皮肤综合征疾病,神经纤维瘤病、结节性硬化、遗传性出血性毛细血管扩张症、共济失调性毛细血管扩张症、脑面血管瘤、VHL综合征。

第一种:神经纤维瘤病,其主要分为2型,分别为神经纤维瘤病I型(NF1)和神经纤维瘤病II型(NF2)。
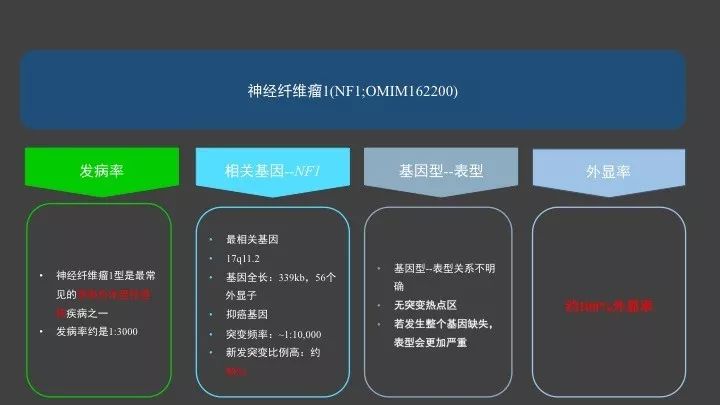
神经纤维瘤1型是最常见的常染色体显性遗传疾病之一,发病率约是1:3000。
与神经纤维瘤1型疾病最相关的基因是NF1,NF1基因属于抑癌基因,全长339kbp,发生致病突变,导致基因产物神经纤维素结构和数量上的改变,从而使其抑癌功能减弱或丧失,导致肿瘤的发生。NF1基因突变频率(~1:10,000),也是人类基因中,突变发生频率最高的基因之一。NF1基因如若发生致病突变,其外显率可达到100%。
在所有已知检出的NF1基因突变的患者当中,NF1基因的新发突变比例很高,约为50%,且在所有NF1相关研究报道中,没有发现突变热点区,也没有相应的基因型和表型相关性的证据,但是有些报道显示,如若NF1发生整个基因缺失,通常会造成相当严重的表型症状。
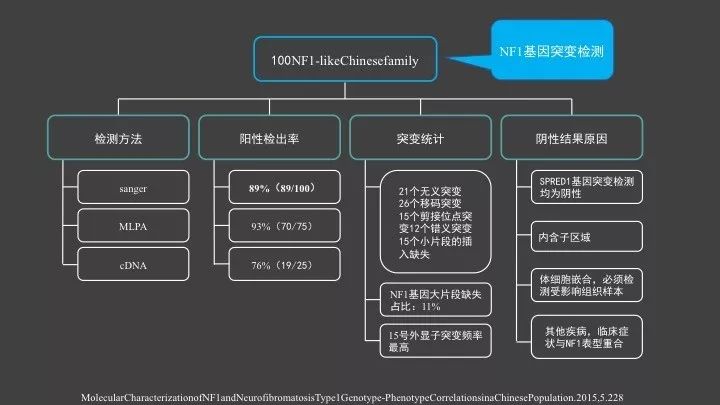
这是发表于2015年,关于NF1基因突变检测的一篇报道,该文章对109个来自于100个家族的临床怀疑NF1型患者进行基因检测,采用的方法是sanger,MLPA,cDNA测序。共在97个来自于89个家族的患者当中发现了NF1基因阳性突变,总体阳性检出率是89%。在符合NIH的诊断标准的NF1患者与只呈现牛奶咖啡斑的患者当中,NF1基因的检出率有明显不同,分别是93%(70/75),和76%(19/25)。
对检出的NF1基因突变进行统计,共发现,21个无义突变,26个移码突变,15个剪接位点突变,12个错义突变,15个小片段的插入缺失。其中,使用MLPA方法共检测到11个片段缺失,总体占比是11%,以往报道的NF1基因片段缺失占的比例在5-10%,与之吻合。在15号外显子中检测出的突变频率很高。
NF1基因突变检测阴性的患者,可能存在以下几个原因:
存在其他基因突变的可能性,目前已知的与NF1相关的基因有:NF1、SPRED1、MSH6、RASA2、KIT、SDHD、PMS2、MLH1、MSH2、SDHB、SDHC等,该篇文章对阴性的患者样本进行了SPRED1基因突变检测,结果均为阴性;
突变位于内含子区域,测序没有覆盖到;
存在体细胞嵌合,若样本为病变组织样本,可能会提高阳性检出率;
其他疾病,临床症状与NF1表型重合,如LEOPARD综合征neurobromatosis-Noonan综合征。

神经纤维瘤病2型的遗传方式也是常染色体显性遗传,发病率约是1:33000,目前已知的与神经纤维瘤病2型最相关的基因是NF2,基因全长114kbp。NF2基因的新发突变比例与NF1基因类似,也是约为50%。
在已报到的NF2患者当中,NF2基因突变,包括点突变和缺失重复,总体检出率约为72%,家系患者检出率较高,为92%。已有研究显示,一些阴性结果的可能原因:存在体细胞嵌合现象,该种情况可能会造成约25%-33%的患者出现假阴性结果,故采用病变组织样本,会提高突变检出率。在所有已知的NF2基因突变中,大片段缺失和重复约占10%-15%,突变范围多在10-600kb。
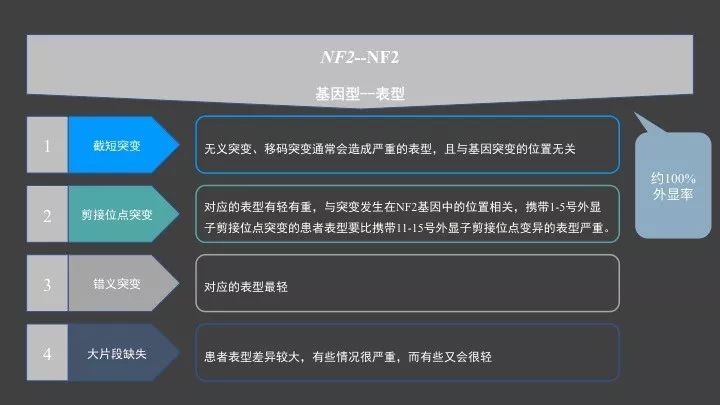
NF2患者当中,同一家族成员之间的表型差异要显著低于不同家族间的差异,这预示着NF2疾病存在着比较明确的基因型和表型的相关性。目前,已知的NF2疾病严重程度与NF2基因突变类型之间的关系已比较明确
无义突变、移码突变通常会造成严重的表型,且与基因突变的位置无关。这种类型的突变通常会引起早期发病,并且会造成NF2相关的颅内脑膜瘤、椎管内肿瘤、周围神经肿瘤的数目增加;
剪接位点突变对应的表型有轻有重,与突变发生在NF2基因中的位置相关,携带1-5号外显子剪接位点突变的患者表型要比携带11-15号外显子剪接位点变异的患者表型严重;
错义突变对应的表型最轻;
NF2基因的大片段缺失导致的患者表型差异较大,有些情况很严重,而有些又会很轻。
携带NF2基因突变的的个体,其外显率接近100%。几乎所有具有生殖系致病变异的个体都会在相应时间内起病,但发病年龄可随变异的类型而不同;
2015年,AdamHexter等发表的一篇文章,研究了1192名NF2患者,NF2基因突变类型与死亡率之间的关系,研究结果与表型严重程度与基因型之间的对应关系的结果类似。

第二种:结节性硬化,包括结节性硬化I型(TSC1)和结节性硬化II型(TSC2)。
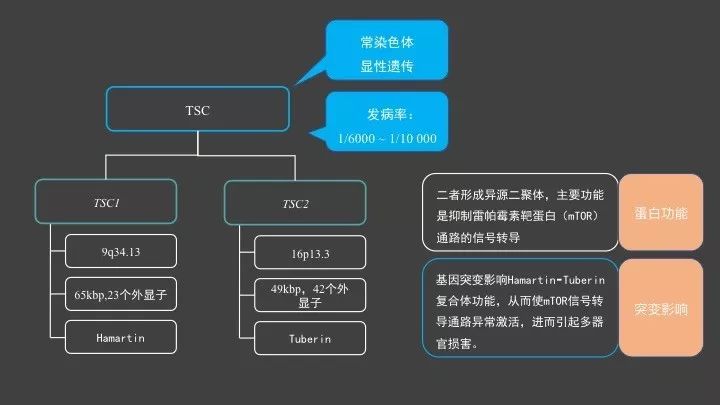
结节性硬化症是一种常染色体显性遗传性疾病,致病基因为TSC1和TSC2,其中TSC1基因定位于第9号染色体(9q34)、TSC2基因定位于第16号染色体(16p13.3)。
TSC1基因含23个外显子,编码蛋白为Hamartin;TSC2基因含42个外显子,编码蛋白为Tuberin。Hamartin和Tuberin具有高度亲和性,二者形成异源二聚体,其主要功能是抑制哺乳动物雷帕霉素靶蛋白(mTOR)通路的信号转导。mTOR在细胞生长和增殖中扮演中央调节者角色。此外,Hamartin和Tuberin还能够单独与多种蛋白质结合,参与一系列细胞生命活动的调节过程,但是其中多种过程的作用机制仍未阐明。结节性硬化症患者TSC1或TSC2基因发生突变,影响Hamartin⁃Tuberin复合体功能,从而使mTOR信号转导通路异常激活,进而引起多器官损害。

起初认为TSC1和TSC2基因突变导致的疾病表型没有太大差别,但是随着研究的不断深入,研究者们发现携带TSC2基因突变的患者表型要比TSC1的严重。散发病例(如一个家族中仅有一名患者)当中,多为携带TSC2基因突变,而TSC的患病大家系中,TSC1和TSC2出现频率几乎相同。
在临床诊断符合TSC诊断标准的患者当中,对应基因突变的总体检出率是在75%-90%,在所有已鉴定的与TSC相关的致病突变中,TSC1基因突变约占20-30%,TSC2致病突变约占70-80%。
在所有已知检出的TSC基因突变的患者当中,新发突变比例很高,约为2/3。在经过对携带TSC1和TSC2致病基因突变的个体统计之后,发现携带TSC致病基因突变的个体,其外显率接近100%,几乎没有携带致病突变而不发病的报道。

第三种:遗传性出血性毛细血管扩张症(Hereditary hemorrhagic telangiectasia,HHT)。
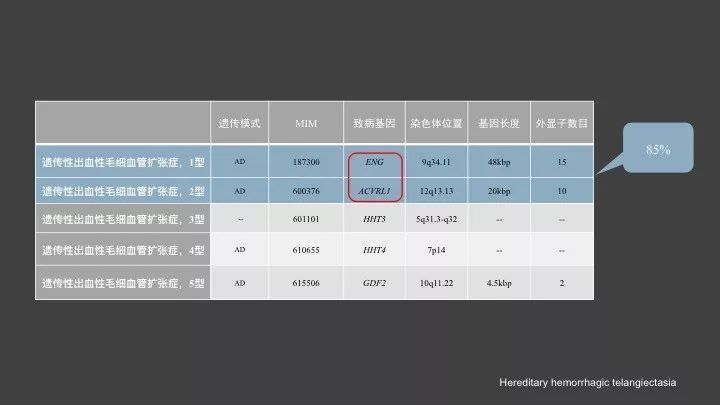
OMIM中报道与遗传性出血性毛细血管扩张相关的基因目前有5个,分别对应5个不同的分型。遗传性出血性毛细血管扩张的1/2/4/5型,对应的遗传模式已经明确,是常染色体显性遗传,遗传性出血性毛细血管扩张3型的遗传模式还未明确。
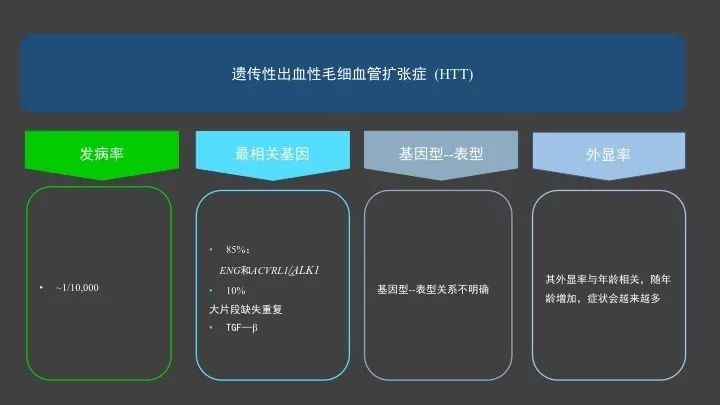
遗传性出血性毛细血管扩张症的发病率约是1/10,000。但是在所有已报道的与遗传性出血性毛细血管扩张相关的基因报道中,ENG和ACVRL1/ALK1两个基因,占了总共的85%左右,而突变类型为大片段缺失和重复的突变比例,分别占这两个HTT疾病相关基因的10%左右。
ENG和ALKl均编码转化生长因子β(transforming growth factory一β,TGF—β)家族的受体蛋白:ENG蛋白和ALK蛋白,这两种受体蛋白主要在血管内皮细胞表面表达,在调节内皮细胞的增殖、分化、粘附、迁徙及细胞外基质的组成和构建上起着重要的作用。因此,ENG基因和ALK基因的突变导致了TGF一β受体蛋白的突变,影响了TGF—β介导的信号通路的正常传导,从而导致血管发育不良而出现HHT。
现有数据和报道显示,对于遗传性出血性毛细血管扩张症的患者来说,没有明确的基因突变与临床表型相关性的支持证据,因为在现有报道中,同一个患病家系中,即使携带相同的基因突变,临床表型也可能会存在很大不同。

第4种:共济失调性毛细血管扩张症(Ataxia-telangiectasia,A-T)。
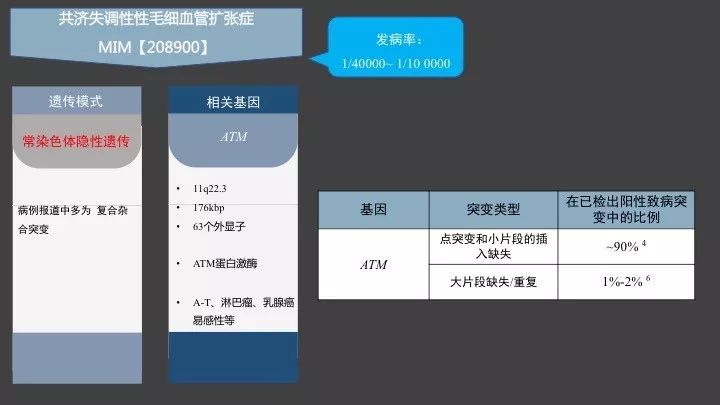
共济失调性毛细血管扩张症,发病率约1/4万至1/10万,其遗传模式与其他常见的神经皮肤综合征疾病不同,属于常染色体隐性遗传。目前已知与该疾病最相关的基因是ATM,全长176kbp,编码ATM蛋白激酶,在DNA的损伤修复当中起重要作用。ATM蛋白激酶的活性与A-T患者表型的严重程度相关,ATM蛋白激酶活性完全消失的患者会出现典型的A-T症状,而具有ATM激酶活性未完全消失的患者,表型会比较温和或非典型。
ATM基因突变除和A-T相关之外,还与淋巴瘤和乳腺癌的易感性相关。目前已知的ATM基因突变共有近千种,且报道A-T患者的病例报道中,复合杂合突变占多数,这种情况下,父母双方各是一个杂合突变的携带者,而现有报道显示,A-T患者的母亲,如果是ATM基因杂合突变携带者的话,患乳腺癌的风险会明显增高。
A-T患者中,目前在已检出阳性致病突变中的患者当中,ATM的点突变和小片段的插入缺失约占90%左右,大片段的缺失/重复约占1-2%。

第5种:脑颜面血管瘤综合征(Sturge-Weber Syndrome,SWS)。
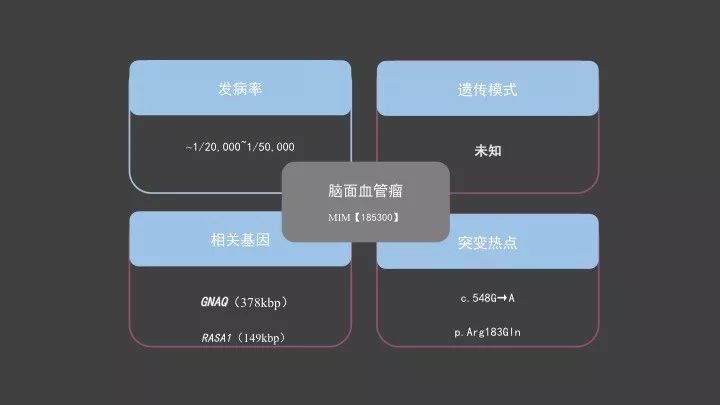
脑面血管瘤,发病率是在~1/20,000~1/50,000。与我们前边所介绍的其他常见神经皮肤综合征疾病不同,对于SWS疾病来说,目前OMIM中并没有明确的遗传学证据显示其对应的遗传模式,但是对于一些具有家系遗传的病例报道中,确实有发现常染色体显性遗传和常染色体隐性遗传的报道。目前关于SWS的基因检测报道相关的文献要明显少于以上疾病,现已知的基因突变多是由基因的体细胞嵌合突变引起,所以这就涉及到基因检测样本的选取,对于该种疾病,一定要取到患病的组织样本和对照才可进行相应的基因检测。
目前已知的与SWS相关的体细胞嵌合基因突变包括GNAQ和RASA1。
目前有一个鉴定比较明确的与SWS疾病相关的体细胞突变的突变热点:GNAQ基因的c.548G→A,p.Arg183Gln。有篇研究报道显示,在对15个SWS患者的血液和患病组织成对样本进行测序之后,共在12名患者的组织样本当中检测到了c.548G>A 的体细胞突变,出现频率是80%(12/15),另一篇样本量数为26个SWS的报道,该位点的检出率是在88%(23/26)。同时该病对应的基因突变有另一特点,就是体细胞突变的频率很低,在这篇15个样本量的研究报道中,该变异位点的突变频率分布是在3.6-8.9%。在未检测到该位点变异的其他3名患者当中未发现GNAQ基因的其他位置的变异信息。这些结果都预示着c.548G>A 该位点可能是SWS患者当中主要的基因变异因素,同时也存在着其他基因突变导致SWS的可能性。

第6种:VHL综合征(Von Hippel-Lindau Syndrome,VHL)。
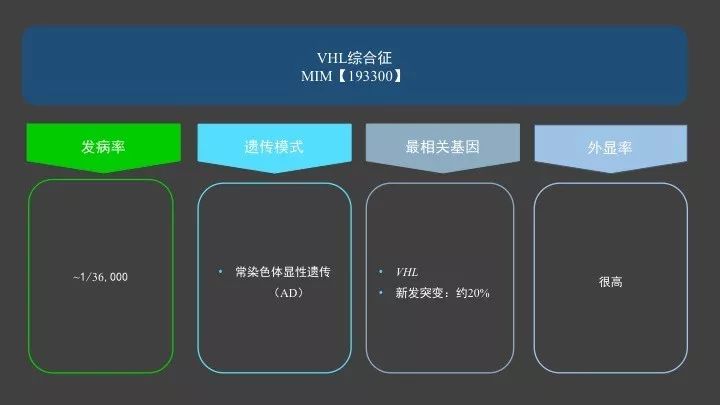
Von Hippel-Lindau(VHL)综合征是一种较为罕见的常染色体显性遗传疾病,可引起包括中枢神经系统在内的多系统肿瘤.VHL基因是一种抑癌基因,VHL综合征由VHL基因突变引起,VHL基因通过促进缺氧诱导因子1α(HIF-1α)降解导致疾病发生,它通过编码VHL蛋白来调控其mRNA,在缺氧条件下,导致血管内皮生长因子过表达,影响肿瘤的生长浸润。除VHL 基因之外,其调节的基因-细胞周期素 D1 基因CCND1的变异也可致 VHL 综合征的发生。VHL综合征中,denovo 突变的比例要明显少于NF和TSC,在已报道的文献中显示,VHL综合征的患者有约80%的个体的致病突变是遗传自患病的双亲之一,仅有20%是denovo突变。
VHL基因致病突变的外显率是特别高的,几乎凡是携带VHL基因致病突变的个体在65岁之前均会出现症状。
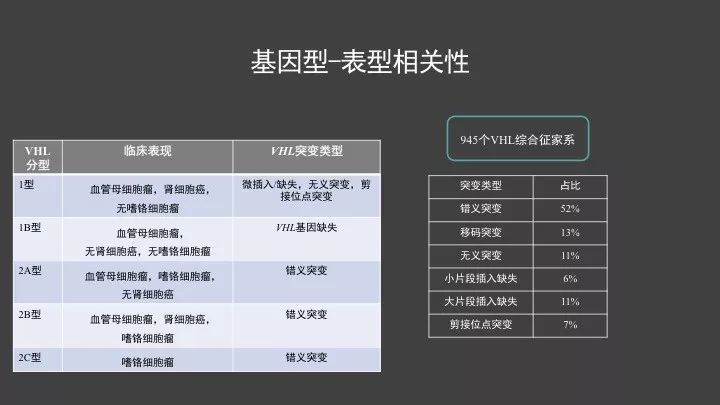
VHL 综合征表型可分为两型:1 型 (无嗜铬细胞瘤),2 型(有嗜铬细胞瘤),其中 VHL2 型又分为3 个亚型,2A 型有嗜铬细胞瘤和 CNS 血管母细胞瘤,不伴 RCC(血管母细胞瘤和肾细胞癌 (renal cell carcinoma,RCC) );2B 型伴 RCC;而 2C 型仅有嗜铬细胞瘤。VHL 综合征1型主要是与VHL基因的微缺失/插入、缺失、无义或剪切位点突变有关,VHL2 型主要是和VHL基因的错义突变有关。
2010年的一篇报道中,对945个VHL综合征的患病家系进行了测序,突变谱分别是错义突变-52%,移码突变-13%,无义突变-11%,小片段插入缺失-6%,大片段插入缺失-11%,剪接位点突变-7%。许多研究证实,VHL疾病的嗜铬细胞瘤的症状特征比起该疾病出现的其他症状,与基因变异之间更具有相关性。该篇研究显示,VHL综合征中症状有嗜铬细胞瘤的患者有83.5%携带VHL基因的错义突变,而症状无嗜铬细胞瘤的家系中携带截短突变的比例要高于错义突变。
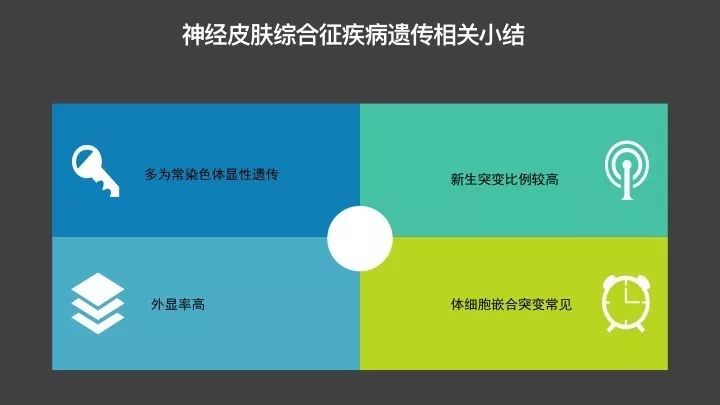
以上就是关于6种神经皮肤综合征基因突变相关研究的一些简介,神经皮肤综合征类疾病,除去一些特殊情况之外,在分子遗传学中有以下4个特征:
遗传模式多为常染色体显性遗传;
检出致病基因突变中,新生突变的比例很高,比如NF为~50%,TSC为~67%等;
致病基因突变的外显率非常高,好多能达到100%,如NF、TSC,VHL综合征等,这种情况下只要个体携带这些基因的致病突变,基本上就会患病;
体细胞嵌合突变在神经皮肤综合征中很常见,这个也是该类疾病的有些血液样本检测不到基因突变的一个重要原因,所以对于这一类疾病来说,如果能提供患病组织样本,将会提高阳性突变的检出率。

因为神经皮肤综合征类疾病的遗传模式多为常染色体显性遗传,所以今天我们主要讲一下常染色体显性遗传类疾病的遗传咨询。
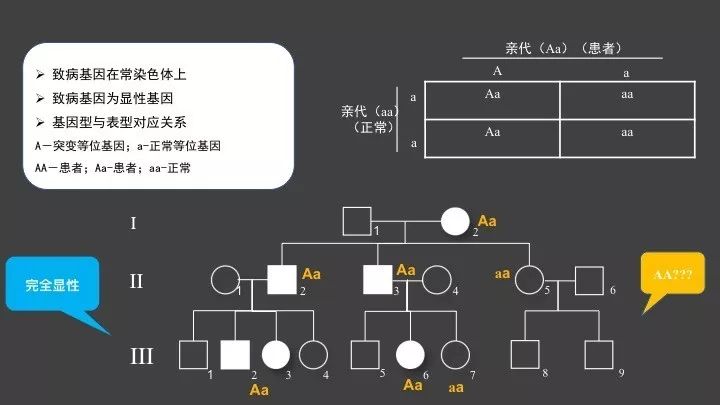
常染色体遗传病是指决定该病的基因位于第1—22号常染色体上,患病与性别无关。常染色体显性遗传病(Autosomaldominant--AD):等位基因之一突变,杂合状态下即可发病。
在常染色体显性遗传病的家系中最常见的是一个患者和一个正常人之间的婚配。假设显性致病基因为A,隐性正常基因为a,最常见的常染色体显性遗传病患者的基因型则为Aa,因为只有当父母均是该遗传病患者,才有1/4的可能生出AA型子女,而致病基因的携带频率一般都很低,所以说这种婚配机会在现实生活中很难看到,所以常见的基因型是杂合型Aa。

对于已确定常染色体显性遗传的先证者父母的基因突变情况,最主要的包括2种,一是父母之一为致病突变的携带者,将致病基因遗传给了后代,二是出现了denovo突变,父母该基因检测未见突变位点,但孩子出现了新发突变。

若先证者的双亲拟再生育,其生育健康小孩的概率也必须建立在先证者致病基因突变已明确,父母对应的基因位点也明确的基础上进行。
若检出先证者双亲之一为致病突变携带者,这种情况下,将致病突变遗传给后代概率是50%。若双亲未携带致病突变,父母双方均为正常个体,再生育患病小孩的概率很低,但会高于普通人群,因为不能排除生殖细胞嵌合的可能性。

如果是该名患者与正常未携带突变的伴侣生育后代,其将致病突变遗传给后代概率也是50%,但是如果生育了携带致病突变的子代,其是否患病和疾病的外显率相关,如NF1,外显率100%,即等位基因之一只要携带突变就会患病,但是疾病表型的严重程度可能不同。

以上就是本次《几种常见神经皮肤综合征相关疾病基因突变研究简介》课程,包括神经纤维瘤,结节硬化、脑面血管瘤、VHL综合征、遗传性出血性毛细血管扩张症、共济失调性性毛细血管扩张症疾病的基因突变研究简介,和常染色体显性遗传类疾病的遗传咨询。感谢各位老师在百忙之中抽出时间收听我们这次课程。
现场提问
1.什么时候会考虑生殖细胞嵌合现象?
答:新发突变的遗传变异可以来自父母也可以是体细胞突变。在某些情况下,突变发生在人的卵细胞或精细胞中,但不存在于任何其他细胞中;在其他情况下,在卵细胞和精细胞结合后不久,在受精卵中发生突变,当受精卵分裂,每个生长的胚胎中的细胞都会有突变。当一个家系中,同一代出现多名相同疾病的患者,但是父母的血液样本筛查检测结果为阴性,这种情况下,我们就需要考虑生殖细胞嵌合现象了。
2.对于这类疾病推荐什么基因检测方式?
答:神经皮肤综合征类疾病突变类型多为点突变为主,大片段的插入缺失所占比例不高,所以从患者经济学考虑的话,可以优先选取以检测点突变为主的测序方式,比如全外显子组测序或是基因panel,如果基因检测结果是阴性的话,再考虑检测大片段的插入缺失的MLPA或是基因芯片等。
3. 所以说对于这类疾病的基因送检样本,最好是病变组织样本?
答:由于存在的体细胞突变嵌合现象,如果送检血液样本的话,可能会由于嵌合比例太低,导致血液样本检测不出,所以病变组织样本会明显增加阳性致病突变的检出率。
