

赛福助力|揭秘同义突变和深度内含子变异导致遗传性癫痫
贵阳市儿童医院儿科神经内科专家通过对15例临床表现高度怀疑遗传性癫痫,但基因未确诊患者的家系全外(trio-WES)数据进行回顾性分析,发现了两个疑似异常剪接事件,进一步通过功能验证实验,揭示了两例遗传性癫痫患者的遗传学病因。赛福基因参与了该项研究工作的数据分析、功能验证实验等临床科研转化工作,研究结果共同发表于BMC Medical Genomics (影响因子 3.8)。

研究背景:
全外显子组测序(WES)方法在检测癫痫患者的致病变异方面非常成功,有研究表明,对于单基因遗传病,trio-WES的阳性诊断率高达28-55%。然而,由于一些其它的因素,如:生物信息学预测流程不完善、变异解释不准确或致病性证据不足,使得检测到的变异难以明确其致病性,从而导致一些临床高度怀疑的的遗传性癫痫无法得到基因确诊。
长期以来,异常剪接被认为是罕见遗传疾病的主要原因。RNA前体(Pre-mRNA)的剪接依赖于外显子-内含子剪接位点、调控序列的精确识别,以及剪接体snRNP与剪接因子的相互作用,任何错误和不准确的过程都可能导致异常剪接。
深度内含子变异可能会导致新的剪接位点的产生,进而导致无义介导的mRNA降解(nonsense-mediated mRNA decay, NMD)或截断蛋白;外显子区域的同义突变也可能通过引入新的剪接位点、激活隐蔽剪接位点或中断外显子剪接增强子来影响剪接。
深度内含子变异通常无法被WES检测到;外显子区域的同义突变也很容易由于软件预测的无害性而在数据分析过程中被忽略。
在本项研究中,通过对2019年7月至2020年6月收集的15例临床表现高度怀疑遗传性癫痫(Dravet综合征,DS;遗传性癫痫伴热性癫痫发作,GEFS+;良性家族性新生儿癫痫,BFNE),但基因未确诊患者的家系全外(trio-WES)数据进行回顾性分析,发现了同义突变和深度内含子变异两个异常剪接事件,进一步通过体外minigene实验证明这两个异常剪接事件可能是未确诊疾病的遗传学原因。
案例回顾:
基因检测:
家庭1:基因检测结果回顾性分析发现患者在KCNQ2基因上存在一同义突变NM_172107.3:c.1617_C > T(p.Ser539=),该变异根据ACMG被判读为“临床意义不明(VOUS)”。应用sanger测序扩大家系验证,结果表明该变异遗传自与先证者表型相似的父亲,且临床表型相似的祖母、姑姑 、堂兄弟(图1A),而无表型的母亲、同胞姐妹及爷爷在该位点则表现为野生型。
家庭2:WES数据回顾性分析未发现可疑变异,但根据患者表型,临床高度怀疑SCN1A基因为其致病基因。根据以往该基因的文献报道,针对SCN1A基因的23号内含子设计特异性扩增引物,并进行sanger测序。测序结果发现,23号内含子中存在一变异NM_001165963.2:c.4002 + 2461_T > C。该变异经SpliceAI软件预测,显示下游28 bp的供体增益为0.18,上游35 bp的受体增益为0.18,表明存在内含子滞留。应用sanger测序扩大家系验证,验证结果表明该变异遗传自与先证者表型相似的父亲 ,且此变异在该家系中共分离(图1B)。
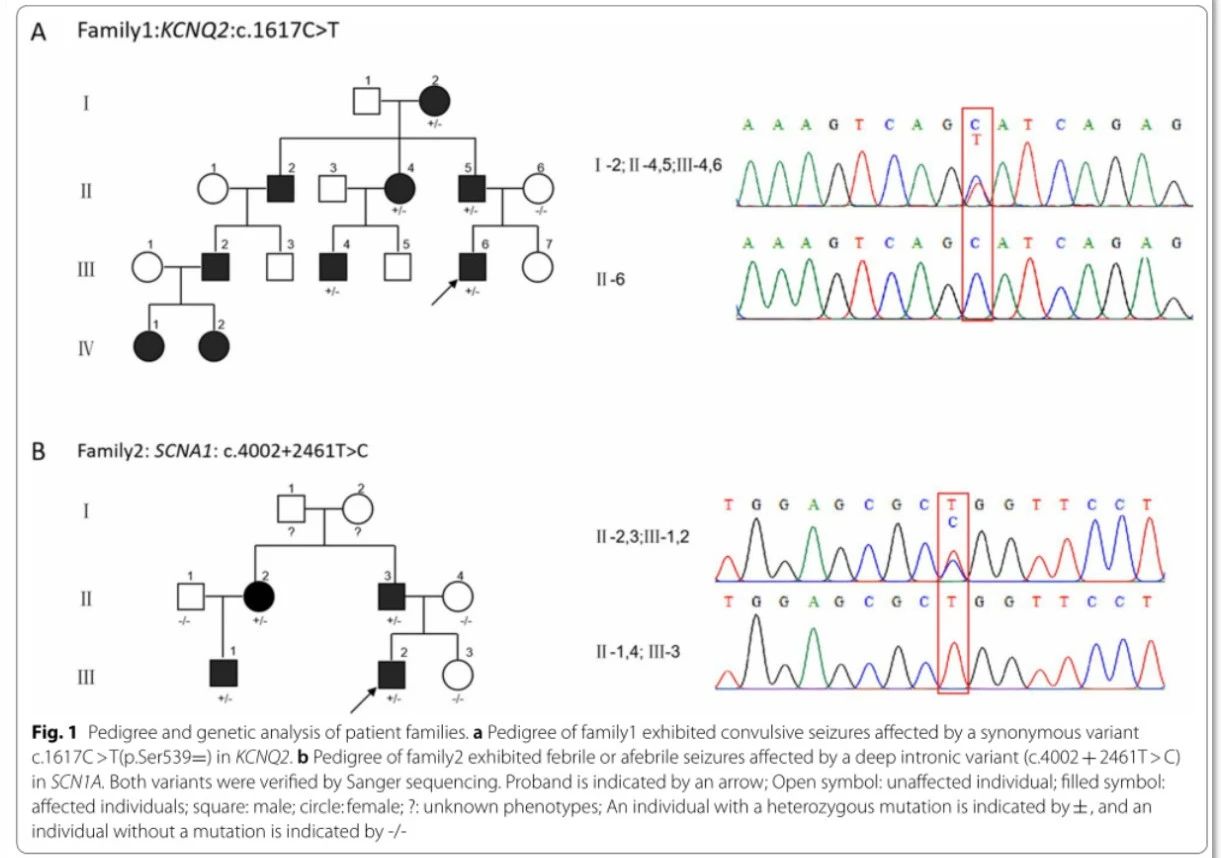
图1 患者遗传家系图谱
功能验证:
KCNQ2:c.1617_C > T变异体外实验结果:体外构建minigene载体,转染人细胞系Hela和HEK-293T,RT-PCR分析显示,与野生型相比突变体显示剪接异常;测序结果显示,该同义突变由于激活了14号外显子中的一个新的隐蔽的5 '供体剪接位点,导致了更短的转录本和20 nt的缺失(图2A, B),该转录本预测会产生一个截断蛋白(p.Val537Cysfs*39)(图2C)。
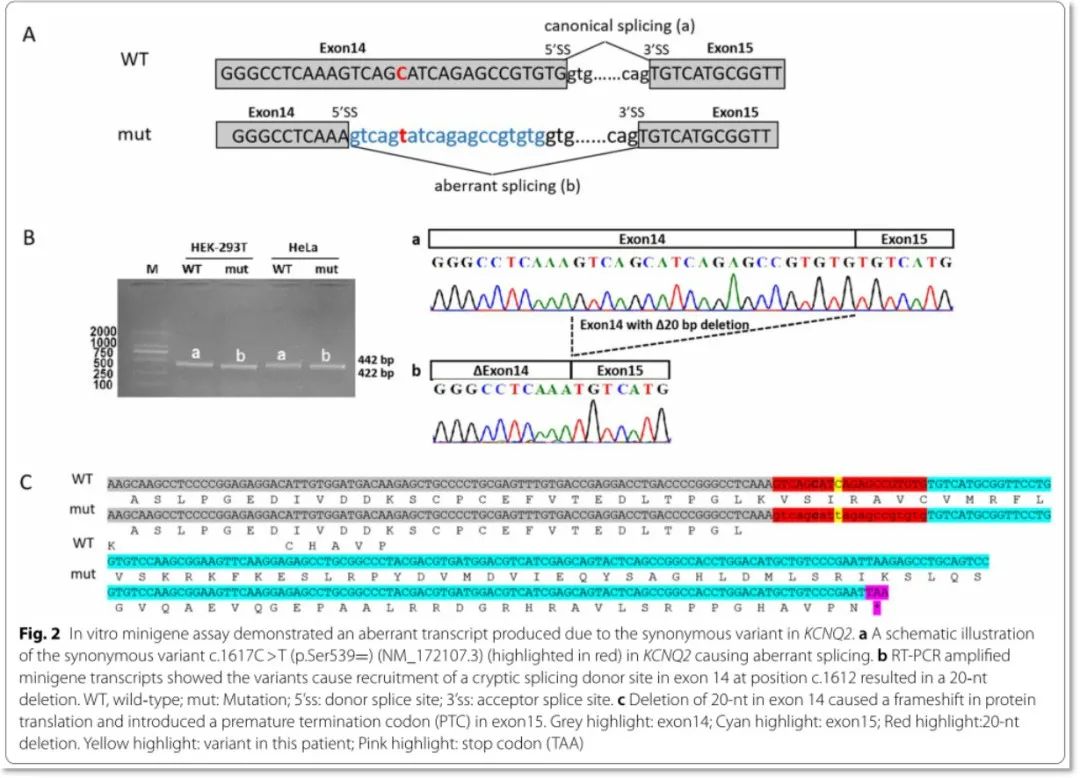
图2
SCN1A:c.4002 + 2461_T > C变异体外实验结果:体外构建minigene载体,转染人细胞系Hela和HEK-293T,RT-PCR结果显示突变体的产物较野生型长;测序结果表明变异体激活了隐蔽剪接位点,导致64 nt内含子滞留(图3);预测64 nt片段的插入将在变异后的65位氨基酸位置导致翻译终止(p.Val1294Aspfs*65)(图4B)
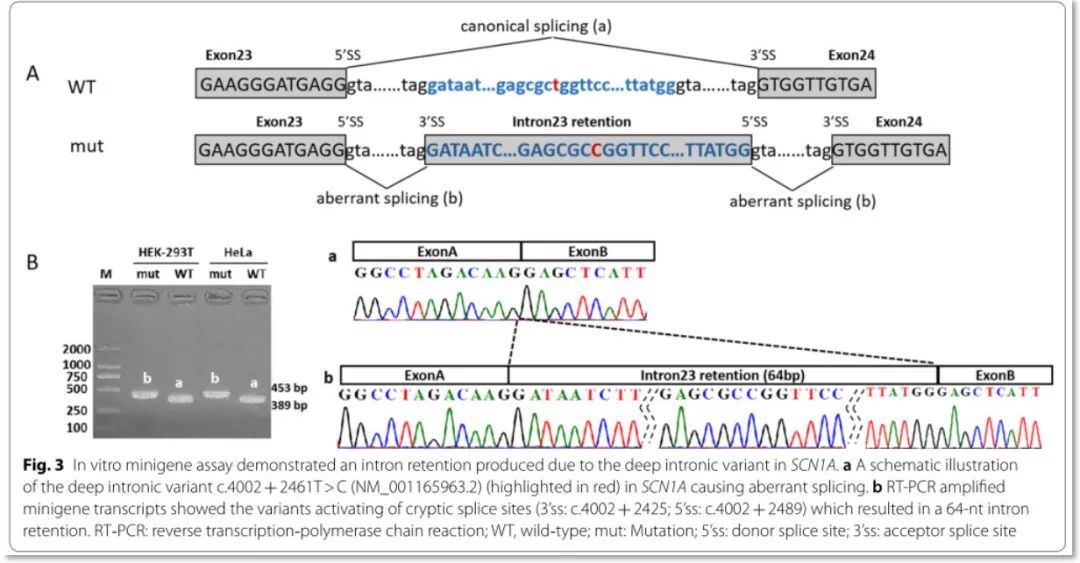
图3
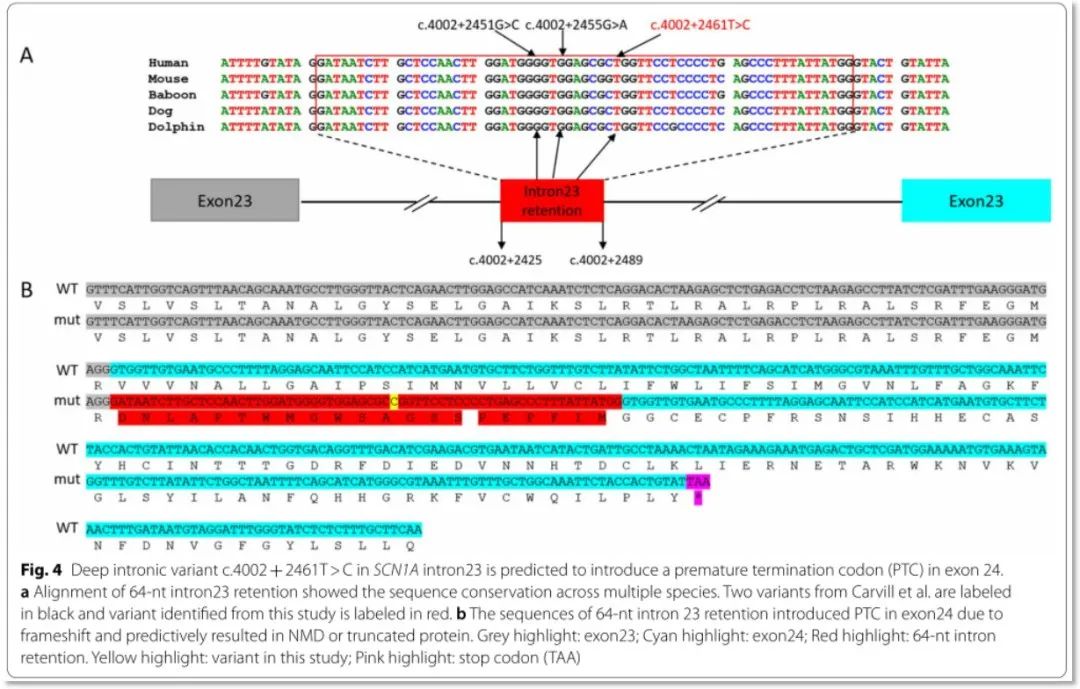
图4
讨论:
有研究表明,孟德尔疾病中约9-30%的致病变异是异常剪接导致的,然而,在人类基因突变数据库 (HGMD) 中收录的所有突变,只有 8.6%是剪接突变(HGMD数据库,更新至2020年6月),因此,影响剪接的同义突变和深度内含子变异的综合研究可对提高罕见病患者的诊断率大有裨益。
在本项研究中,我们报告了两个WES遗漏的重要变异:
(1)SCN1A:c.4002 + 2461_T > C由于其位于深度内含子而未被外显子组测序发现;
(2)KCNQ2:c.1617 _C > T被常规生信分析流程忽略;
我们通过进一步的功能实验揭示了这两个异常剪接事件是未确诊疾病的遗传学原因。
以上案例提示我们,对于那些有家族史,且临床高度怀疑是由遗传因素导致的病例,在WES阴性的情况下,应特别注意一些容易被忽略的同义突变/深度内含子变异,并结合生物信息学软件和功能实验来揭示未确诊疾病的遗传学病因。
来源:BMC Medical Genomics
原文链接:Unraveling synonymous and deep intronic variants causing aberrant splicing in two genetically undiagnosed epilepsy families